
Micro-device could pick up early signs of heart attack or stroke
Heart attacks and strokes are the world’s leading cause of death

Heart attacks and strokes are the world’s leading cause of death

Oestrogen may protect diabetes patients from cardiomyopathy, according to research led by a Texas A&M AgriLife scientist and published in the June issue of the American Heart Association journal Circulation: Heart Failure https://doi.org/10.1161/CIRCHEARTFAILURE.121.008758

Levels of bad cholesterol rise during menopause, and 10% of this increase is due to shifts in sex hormones. That’s the finding of research published in May in the European Journal of Preventive Cardiology, a journal of the ESC. [1]
Recommendations on the prevention and management of interference caused by medical procedures in patients with implanted electronic cardiac devices has been published in EP Europace, [1] a journal of the European Society of Cardiology (ESC) and presented at EHRA 2022, a scientific congress of the ESC.
When should patients and family members undergo genetic assessment for a heart condition? An international consensus document published in EP Europace, [1] a journal of the European Society of Cardiology (ESC) and presented at EHRA 2022, a scientific congress of the ESC, sets out the recommendations.
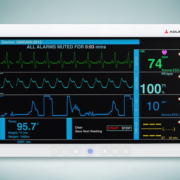
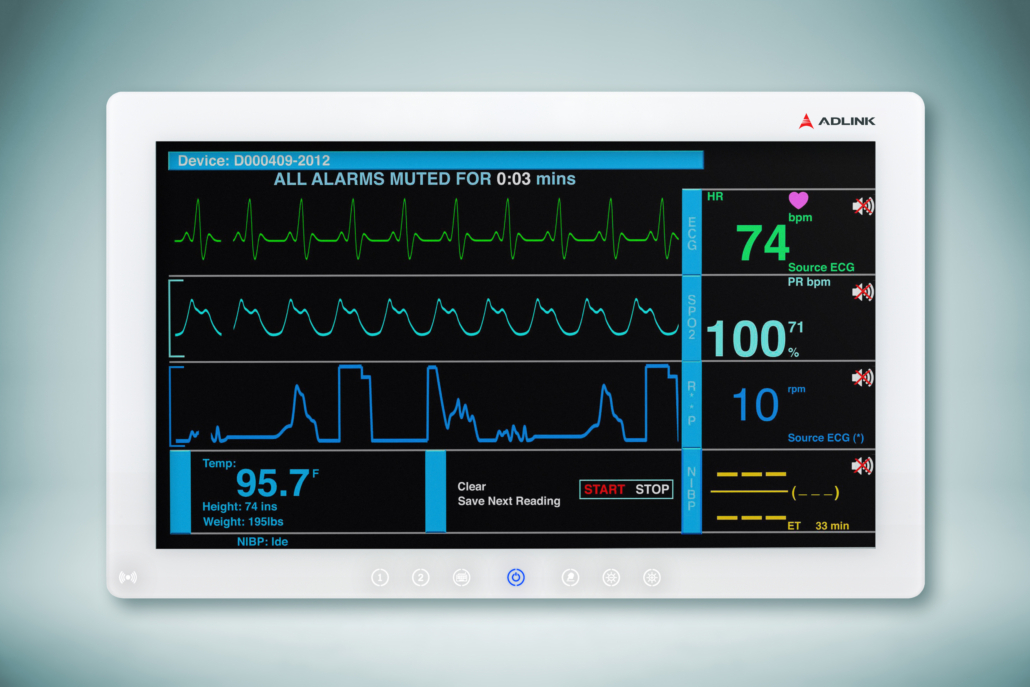
Choosing the best medical PC for operating room use is an important decision for hospitals around the globe. Thankfully, advances in medical PC design and operation have lifted their hygienic-friendliness to an impressive level that is welcomed by healthcare professionals. This article will take a look at how medical PC design has changed and what factors should be considered for your next purchase.
For many years, plastic was used to cover PC monitors and keyboards in operating rooms to ensure hygienic use. It was then removed, thrown away and replaced with fresh plastic after each use. The utilitarian design and construction of PCs with various cracks, crevices and attachment points did not take well to hygienic cleaning and disinfection. Sensitive monitor screens could not withstand aggressive cleaning procedures and keyboards hid dirt and debris between the keys, which made them extremely hard to clean and disinfect.
Hospitals stepped away from these legacy systems when medical PC manufacturers started producing systems specifically designed for hospital use. The latest systems are designed with cases that are smooth, sealed, free of vents or fans, and come with Class 1 medical device approval. Interfaces are also completely sealed and come with IP protection to prevent cleaning and disinfectant liquids from entering the case and damaging electronics.
Although the hygiene of PCs was always important for hospitals, the current pandemic has made hygiene more crucial than ever before.
Although the threat of nosocomial infection in hospitals cannot be compared to the current pandemic, it is still an important consideration.
For example, patients with infections acquired from intensive care units in developed countries range from 3.5% to 12%, with a frequency rate up to 17 episodes per 1000 patient-days. Undeveloped countries have a range from 4.4% to 88.9%, with a frequency up to 42.7 infections per 1000 patient-days. This gives us a total of 6.2 infections per 365 patient-days in developed countries and 15.58 infections per 365 patient days in undeveloped countries. These infections account for 37,000 deaths in Europe and 99,000 deaths in the USA.
Compared to 207,784,507 infections and 4,370,424 deaths worldwide due to the pandemic as of 17 August 2021, infections in hospitals seem to be very minor. But even after the exit of the pandemic, these infections will still be with us. Therefore, reducing health-care related infections is, and will remain, a major task for hospitals around the globe and one that must be addressed to help save lives.
One new approach is the use of UV light as a disinfectant. UV-C light, specifically, is being used in operating rooms for reducing germs and viruses. Research carried out by Dr. Anthony Griffiths, Associate Professor of Microbiology at Boston University School of Medicine, has shown that viruses exposed to a radiation strength of 0.849 mW/cm2 at a level of 7.64 mJ/cm2 for just nine seconds can render them inactive. It is a good way to sterilize the air in an operating room, or disinfect smaller devices, but it is not fully effective when eliminating pathogens on larger medical devices like medical PCs. Therefore, the process of physical cleaning and disinfection in these cases is still necessary.

Dirt accumulates in computer vents
Advances in PC technology have made it possible to design high-performance PCs that are completely sealed, which means no vents or fans that could serve as agents for germ distribution. Although this passive cooling design is extremely advantageous for cleaning and disinfection, it limits the PC’s performance to a maximum of 150 watts. Medical PC manufacturers must therefore incorporate components and operations that deliver the highest performance possible without crossing the 150 watt border. Thanks to sophisticated designs and highly efficient embedded components, today’s medical PCs provide enough computing performance to even render high resolution 3D images during operations, while remaining very energy efficient, which is very advantageous for use on patient care carts that utilize batteries for power.
A sealed, passively-cooled case is an important part of the hygienic PC formula, but is not the only thing to consider. An equally important component is the display panel. Today’s best design principle for medical PCs is the use of touchscreens that feature edge to edge coverage, which eliminates the gaps that normally exist with screens recessed in the housing. The result is a display that is much easier to clean and disinfect on a regular basis.
It’s crucial to use robust glass for the touchscreen, so it holds up to aggressive cleaning and disinfection without scratching or dulling. The glass surface should also be antireflective to reduce light source reflections that could interfere with the clear display of images. The best solution is to use etched glass with a minimum hardness of 7H, instead of applying a sensitive antireflective coating to the glass that is prone to scratches and wear and can lose its effectiveness over time. And lastly, keep in mind that the touchscreen should be sensitive enough to recognize touches with surgical rubber gloves.
Although a completely sealed case is important for hygiene, it should also be extra sturdy and offer a high level of impact-resistance in the sometimes fast-paced and hectic operating room environment. As the PC system is normally mounted on a flexible arm in operating rooms, it’s exposed to bumps and hits during use, such as by a moveable light. The case should therefore withstand a 250 g drop from a 40 centimeter height, which corresponds to an impact resistance level of IK06.
An active antibacterial coating on the surface of the case adds to the hygienic design. Such coatings contain silver, which provides an effective level of protection against pathogens and should have an effective service life of more than five years.
A recent innovation that contributes to an even higher level of hygiene is invisible housing connections that were developed and patented by Adlink Technology. With the use of this connection system, hard to clean cracks, crevices and low-lying screw heads make the cleaning and disinfection process not only more effective but also quicker and easier.
Instead of using screws, the case panels are magnetically attached. A spring-loaded latch holds the case together and does not feature any external components. The latch could be designed, for example, as a pin that can slide into place for attachment of the touchscreen. Alternatively, it could be an integrated part of the case. Attachment of the touchscreen to the PC base is therefore fully secure. It can only be decoupled intentionally and not by accident.
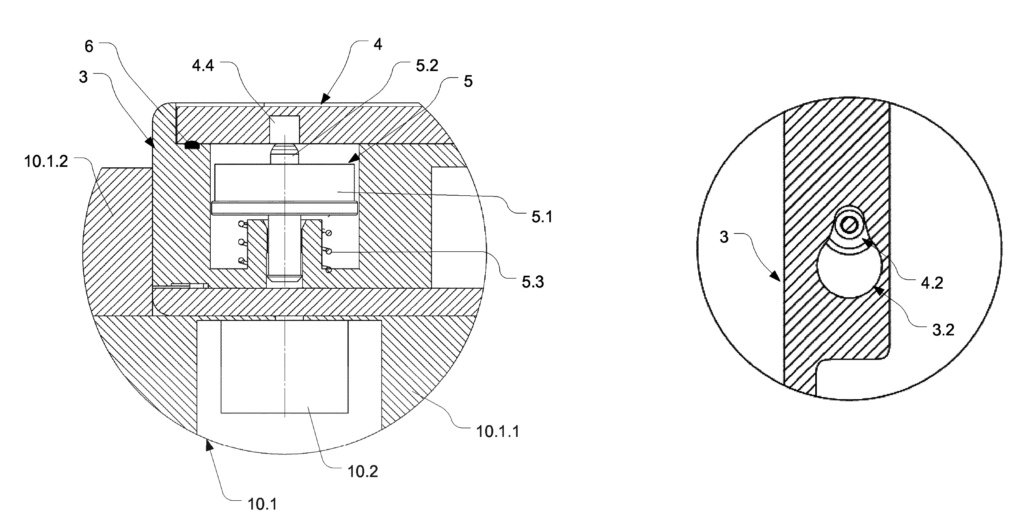
The patented connection system replaces external screws with internal bolts that are magnetically positioned from the outside of the case. The magnetic safety bolt (5), with magnetic piston (5.1) and locking pin (5.2), lock the case cover (3) to the base (4). An external magnet (10.2) pulls the safety bolt against the force of the spring to decouple the two halves of the case. When closing the case, the locking pin automatically goes into the hole (4.4) to secure both halves of the case to each other.
This purpose of this article was to provide the reasons why MDR Class I certification of a medical PC, as well as its observance of EN60601-1 and EN60601-1-2 technical standards, are not the only important considerations when deciding on your purchase. A range of additional criteria should be considered by IT Managers in hospitals and other health-care facilities when assessing the hygienic design and operation of a medical PC to help ensure they make the best choice.
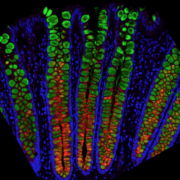
UNC-Chapel Hill scientists sequenced the genes expressed in individual single cells from human GI tracts to discover new cell-type characteristics and gain insights into important cell functions such as nutrient absorption and immune defence.
A new joint guideline published 1 April from the American College of Cardiology, the American Heart Association, and the Heart Failure Society of America, increases the focus on preventing heart failure (HF) in people who are showing early signs of “pre-heart failure,” and updates treatment strategies for people with symptomatic heart failure to include SGLT-2 […]
Megatrends Digital transformation is, without question, one of the most significant revolutionary trends in the history of mankind, comparable to the invention of the first wheel, Gutenberg’s printing press, or the Internet. We are now able to systematically digitize countless information and data that until relatively recently, needed to be stored and locked up in […]

Los Angeles’ Cedars-Sinai’s newest department, Computational Biomedicine, is up and running. International Hospital speaks to the department’s founding chair, Jason H. Moore, PhD., about the history of this relatively new field, how to mine big data and the future of artificial intelligence in healthcare.
Prins Hendrikstraat 1
5611HH Eindhoven
The Netherlands
info@interhospi.com
PanGlobal Media IS not responsible for any error or omission that might occur in the electronic display of product or company data.
